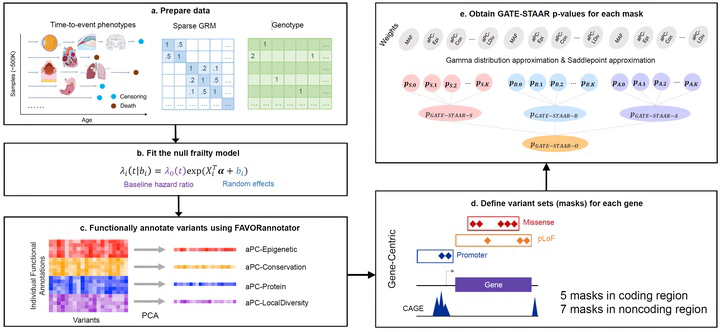
Abstract
Whole genome sequencing (WGS) studies in large biobanks provide an unprecedented opportunity to study the rare-variant (RV) effects on the natural history of human diseases by analyzing censored time-to-event (TTE) phenotypes, such as age at disease diagnosis, disease progression, and lifespan. Unlike existing methods developed for continuous and categorical phenotypes, rare-variant association tests (RVATs) for TTE phenotypes in large biobanks face several major challenges, including heavy censoring, cryptic relatedness, and population structure. We introduce GATE-STAAR (Genetic Analysis of Time-to-Event phenotypes via the variant-Set Test for Association using Annotation infoRmation), a powerful and computationally efficient frailty model framework for RVATs of TTE phenotypes in large biobanks. GATE-STAAR accounts for high censoring rates, cryptic relatedness, and population structure in large biobanks, while incorporating multifaceted variant functional annotations to improve power and result interpretability. We propose a rare-variant saddlepoint approximation method to effectively address heavy censoring in WGS TTE analysis. We demonstrate through extensive simulations that GATE-STAAR is powerful while maintaining proper control of type I error rates. We apply GATE-STAAR to analyze the WGS data of approximately 400,000 UK Biobank participants of white British ancestry across a variety of TTE phenotypes, and validate the findings using participants of European ancestry from the All of Us Research Program. These analyses uncover RV associations with age at diagnosis of a range of diseases.
Type
Publication
Proceedings of the National Academy of Sciences (PNAS)
Significance
Rare variants (RV) identified through whole genome sequencing hold great promise for elucidating the genetic basis of disease onset, but existing methods for RV association testing are not well suited for time-to-event phenotypes. Here, we develop GATE-STAAR, a scalable and accurate framework integrating frailty modeling with functional annotations. We propose a rare-variant saddlepoint approximation to handle heavy censoring. Through comprehensive simulations and large-scale analysis of approximately 400 K UK Biobank participants, with replication in approximately 230 K All of Us participants, GATE-STAAR uncovers biologically meaningful RV associations while ensuring rigorous control of type I error. This framework is powerful to dissect the genetic architecture of disease onset and progression and advance precision medicine.